
Welcome!
We are a computational biology group at the Department of Biomedical Informatics of the Harvard Medical School. We are focused on the analysis of intratumoral heterogeneity in different cancer types, as well as the interactions between tumor cells and their microenvironment. We also study growth and maintenance of healthy tissues, to understand the statistical properties and feedback mechanisms that enable normal biological function.
We specialize in the development of methods for statistical and integrative analysis of the functional state of the cell from high-throughput genomic measurements, with an emphasis in the statistical modeling of the biological tissue from single-cell measurements. The wetlab component of the lab is aimed at automating and scaling up key functional genomics assays using microfluidics.
Wiring together scRNA-seq datasets
scRNA-seq is now applied in complex study designs, which can cover many samples, spanning multiple individuals, conditions, or tissue compartments. Joint analysis of such extensive, and often heterogeneous, sample collections requires a way of identifying and tracking recurrent cell subpopulations across the entire collection. Conos (Clustering On Network Of Samples) is a tool for joint analysis of such collections, that relies on multiple plausible inter-sample mappings to construct a global graph connecting all measured cells. The graph can then be used to propagate information between samples and to identify cell communities that show consistent grouping across broad subsets of the collected samples. Conos results enable investigators to balance between resolution and breadth of the detected subpopulations. Please see the publication for detailed description and analysis examples, as well as the github page for hands-on tutorials.

RNA velocity
Single-cell RNA-seq measurements provide a powerful approach for studying complex biological tissues. Some of the more interesting contexts involve dynamic processes, such as development or disease progression. However single-cell measurements only capture a snapshot of a transcriptional state at a single point in time. To infer dynamics of the cells, together with Sten Linnarsson's group, we have developed a method (velocyto) to estimate time derivative of the transcriptional state for individual cells. This provides basis for quantitative modeling of cell dynamics and the associated regulatory processes.
Exploring Transcriptional Heterogeneity
Single-cell mRNA-seq now allows to measure snapshots of transcriptional state for thousands of cells. Such measurements can be used to explore composition of complex tissues in the context of both healthy homeostasis and disease. Identifying transcriptional subpopulations and features that separate within a given cell population can be challenging, particularly when there are multiple valid criteria on which the cells can be distinguished. For instance, multiple cell types in the mixture may be going through cell cycle and therefore share a very prominent mitosis signature, which may dominate the resulting cell classification. We have developed Pagoda and Pagoda2 for analysis, visualization and interactive exploration of single-cell RNA-seq datasets.
Repression of Fetal Hemoglobin
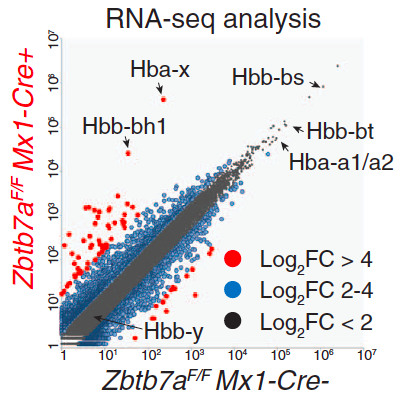
Mutations in adult-type globins underlie several diseases, including sickle cell disease and thalassemia, and are a significant public concern. One promising approach for their treatment would be re-activation of the fetal-type hemoglobin in adult erythroid cells. Fetal-type hemoglobin is normally repressed soon after birth, as the adult-type globin genes take over. The exact mechanism of this repression is not known, though earlier studies have implicated BCL11A as one of the necessary factors. In our collaboration with the laboratory of Takahiro Maeda from the Children's Hospital, Boston, we have investigated the contribution of another factor, LRF, to repression of fetal-type hemaglobin. Through transcriptional and epigenetic analysis we show that LRF acts independently of BCL11A to repress the fetal hemaglobin through direct interaction with these loci. Science [DOI:10.1126/science.aad3312]
Microfluidic devices


The throughput and accuracy of the modern sequencing assays can be improved by performing key steps within automated microfluidic devices. Small volumes allow one to increase the effective concentration of the reagents while reducing sample requirements, and computer-driven control can reduce technical variability.
We are adapting the devices designed by leading microfluidics groups, and working on custom chips to enable high-throughput versions of assays developed by our experimental collaborators.
 National Institutes of Health
National Institutes of Health